

***
FDA Leadership’s “Blind Spots” Lead to a Surge in Medical Device and Drug Recalls
By – brownstone.org
Americans assume that when they pick up a prescription from their pharmacy, an FDA quality control professional has verified that the contents are manufactured to rigorous agency standards. Unfortunately, that assumption is becoming harder to defend.
For decades, the FDA’s drug oversight model has depended on in-person inspections of the manufacturing practice and independent verification of product quality. While no regulatory system is perfect, the underlying principle was straightforward: companies seeking to profit from selling medicine to the American people would be held to strict and independently assessed FDA-enforced standards. However, that principle has recently been eroded with a newly enacted FDA policy.
FDA “Remote” and “Announced” Inspections Began During Covid….But Never Stopped
During Covid, the FDA sharply curtailed – then mostly eliminated – routine in-person inspections as part of its five-year-plus “work at home” policy. The repercussions of that poorly thought-through initiative are still being felt by patients and consumers today. Manufacturers now know that no FDA inspector is going to knock on their doors for an unannounced in-person inspection as they once did. Today, approximately 90% of FDA overseas inspections are announced in advancethrough State Department travel communication requests initiated by the FDA.
Beginning in January 2021, almost immediately following Biden’s presidential inauguration, FDA career employees quickly and quietly proposed an agency-wide “remote” inspection system for almost everything it regulated.

Both the significance and folly of that misguided FDA policy implementation cannot be overstated.
What began as an emergency, temporary Covid-era “work at home” accommodation solidified into a permanent FDA policy. And the result has been a striking failure. Recalls soared and continued to do so even after the FDA ended its “work from home” policy in March 2025, illustrating that underlying remote/announced inspection methodology isn’t as effective as live inspections.
Even with remote testing, right before Trump was elected president in September 2024, the FDA had an inspection backlog numbering in the thousands—and that was just in the US alone. Fast-forward to 2026, and that FDA inspection backlog persists, placing Americans in perpetual danger from unsafe pharmaceuticals and medical devices.
It’s to the point that even the US Government Accountability Office publicly scolded the FDA in February 2026 about not effectively inspecting manufacturing plants.
Today, hardly a weekday goes by at the FDA where they don’t announce at least one recall. This means that the limited number of “remote” drug inspections conducted by the FDA apparently aren’t being conducted effectively.
Recalls may be caused by things such as: contamination issues, falsified records, manipulated testing data, and questionable manufacturing practices that should otherwise have prompted immediate, regulatorily punitive consequences above and beyond just the recall itself.
China and India Manufacturing Quality
A vast majority of America’s pharmaceutical supply chain has moved overseas, with Chinese and Indian manufacturers now producing anywhere from 80 to 90% of active pharmaceutical ingredients. The remaining percentage tends to be brand-name drugs, narcotic, or controlled substances which China isn’t allowed to manufacture by their government, having definitively learned their lesson from the Opium War of the 1800s.
American consumers and patients rely heavily on overseas Indian and Chinese manufacturers, where quality control is widely known to have not only serious shortcomings, but in some cases, deliberate product manipulation, adulteration, and/or fraud. This creates obvious dangers for American patients, particularly when regulators fail to independently orproactively verify quality before products reach America’s pharmacies, hospitals, and patients.
Over the past couple of years, a growing drumbeat of reports has detailed increasingly worrisome findings about overseas drug manufacturing facilities supplying the American market. It began almost immediately after the FDA started working from home and only got worse over time.
It seems that America’s FDA leadership had significant “blind spots” to a surge in product recalls by allowing its “remote” inspection policy to continue as illustrated in the graph below.
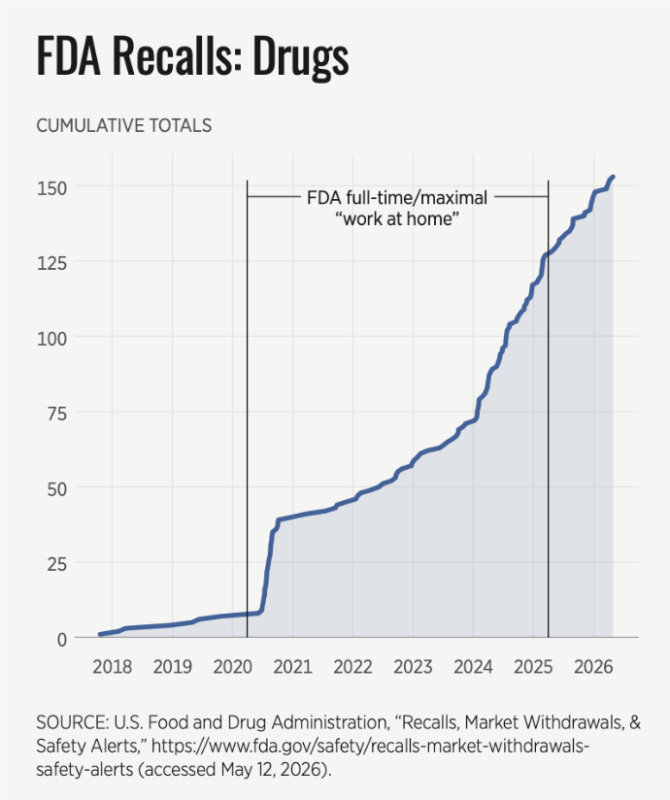
Note: The FDA does not share drug recall data prior to 2018 on its website.
Delayed Messaging to Patients by FDA
Even more concerning than the increasing number of FDA products which made it to market was the time that these recalled products stayed on the market afterwards. Recalled products may have remained available to consumers for weeks or months – but we don’t know, because the FDA does not always publicly detail that reporting delay. Bottom line: American patients are being placed at risk because of an objectively ineffectual FDA inspection regime.
Blind Trust on Complex FDA Devices and Other Products
Drugs are not a product where quality can simply be assumed from a manufacturer’s self-submitted paperwork. Manufacturers are for-profit entities which have every financial motivation to curate their reporting to show the best possible results. When the FDA is reduced to pre-announced, scheduled, and/or remote inspections instead of examining operations firsthand through unannounced visits, the opportunity for quality concealment and risk to the public expands considerably.
When it comes to making drugs, even tiny errors can produce catastrophic consequences. Research has established an estimated 2–3σ (sigma) level of imprecision when it comes to small molecule pharmaceutical manufacturing. That corresponds to 66,807 to 308,537 defects per 1,000,000 opportunities, according to an article published by FDA officials.
But the drugs we take are the most regulated product in the US, and newer and more complex pharmaceutical products including peptides and large injectable biologics, could create more than 1,000,000 “opportunities” for error. A contaminated batch, an improper sterility protocol, or an undisclosed ingredient used to save money on the manufacturing process—causing even one minor structural shift in a molecule—have the potential to permanently injure or kill patients.
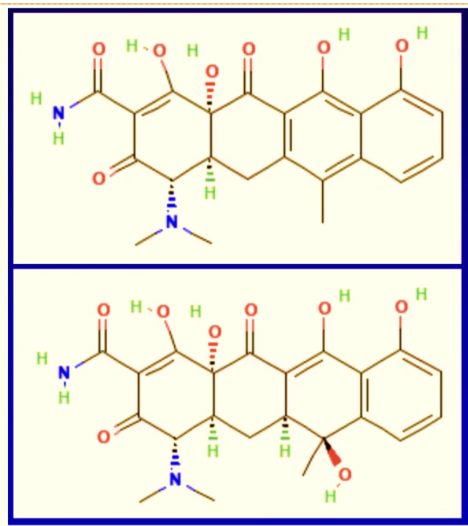
The above image shows chemical stick structures of tetracycline and epianhydrotetracycline. One of these structures is a relatively common, historically safe antibiotic, the other is a poison that can cause renal failure after just two days of dosing. That transformation to toxicity can be caused by a single structural shift from a missing oxygen bond. It illustrates how even very minor errors in manufacturing quality means the difference between a therapeutic drug and a poison.
Any alteration in structure that occurs during manufacturing has the potential to vastly change a compound’s clinical activity, including a change from therapeutic drug into a poison.
FDA-Regulated Device Recalls
FDA Class 1 device recalls—those with the potential to cause serious injury or death—hit a 15-year high in 2022 and continued to balloon in 2024 and 2025.
The FDA device recall page has well over 150 recalls involving thousands of products for patients in 2024 alone. Although the FDA had a dedicated 2024 webpage for device recalls, the FDA stopped generating dedicated webpages for 2025 or 2026, per a rudimentary internet search.
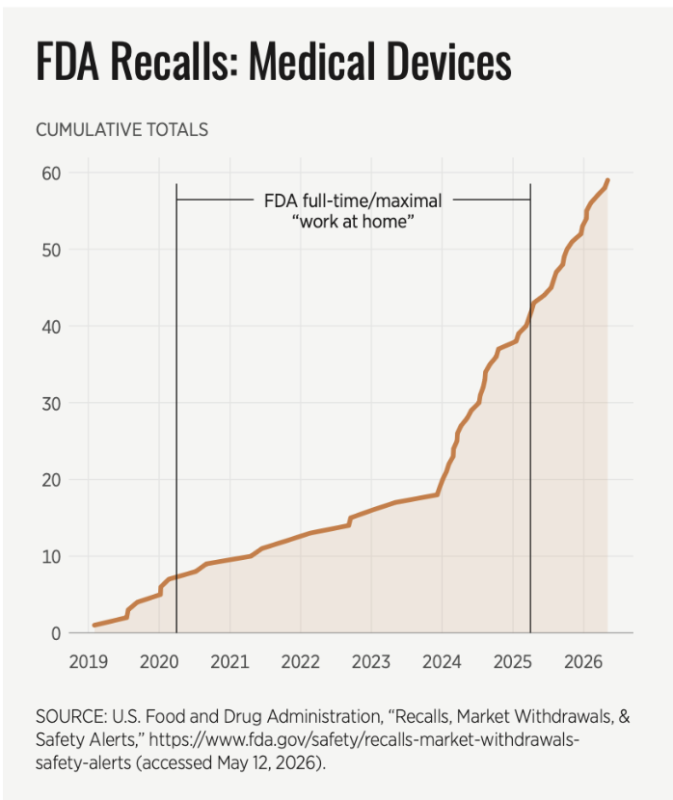
While some of the listed recalls were for things which could seem innocuous like “shortened battery life” if the product in question is a surgical anesthesia system, defibrillator, or a patient ventilator or an extracorporeal membrane oxygenation system, even a “shortened battery life” has the potential to be life-threatening.
FDA devices are a completely different entity as compared to pharmaceuticals. While drug quality regulation essentially focuses on assuring reliable qualitative and quantitative aspects, devices can range from anything from a syringe, to a molded implantable joint, to a hemodialysis system. Devices may heavily rely on a myriad of medical-grade pumps, sensors, complex hardware, synthetic materials, electronics, and software, all of which would collectively need to be verified to have been manufactured to exacting quality specifications to be considered safe.
Unfortunately, the FDA rarely forces manufacturers to recall dangerous medical devices. Instead they wait for device failure reports or deaths from physicians and patients.
In other words, the recalls we are seeing have potentially already resulted in patient harm.
A 2024 Government Accountability Office probe found that in 2022, the FDA received 3 million reports about malfunctioning devices — nearly 30 times more than in 2005. Nearly one-third described injuries and deaths. Still, the FDA doesn’t use its authority to force manufacturers to pull defective devices from the market, even though federal law empowers them to do so.
The Current FDA Inspection Policy Isn’t Adequately Protecting Americans
Despite the high stakes of placing patients at risk, the FDA’s current policy asks the public to trust for-profit drug manufacturers which are primarily inspected during scheduled and announced intervals (as opposed to unannounced visits), which on top of that, do not occur frequently enough, and in most cases, are permitted to be inspected “remotely.”
The agency has also failed to provide adequate notification that the public deserves when quality concerns arise. In many cases, the FDA is aware of recurring manufacturing deficiencies long before the public ever learns of them, yet often provides limited and/or delayed communication regarding which manufacturers repeatedly fail inspections or submit unreliable data.
In addition to not always giving timely notification, the FDA does not adequately inform the public about specific findings of its testing when it does uncover impurities or errors. This information is not eligible to be shared with the public even through FOIA requests. Americans are usually only made aware when the matter escalates into a public relations crisis and is reported by network news.
Summary
In conclusion, Trump’s (now exited) FDA leadership inherited an agency already weakened by pandemic-era disruptions, but it also inherited its historical obligation to enforce rigor to one of the most critical, consequential, and historical functions when it comes to FDA oversight: safety. Well over a year after FDA leadership was confirmed, FDA recalls are climbing and likely will continue to climb, placing America’s patients at grave risk for morbidity and mortality unless a rapid course correction occurs.
Adding to the lack of misguided policy: each year, the FDA conducts approximately 12,000 manufacturing plant inspections in the US, but just 3,000 inspections across 90 other countries that, according to the FDA: “produce foods, essential medicines, and other medical products intended for American consumers and patients.” In addition to unfairly punishing domestic manufacturers with more inspections as compared to those overseas, this policy has not yielded a positive effect on the after-market recalls of drugs and medical devices as illustrated in the graphics shown here.
Drug and device recalls accompany a surge in food recalls. It’s more than a little ironic, because the now-exited FDA Commissioner Marty Makary promised Americans that the safety of our foods would be emphasized during his leadership. He frequently and repeatedly stated that “people forget the ‘F’ in FDA stands for food,” and “we want to focus on food,” and “It’s time to tell people the truth about food.”
The surge in recalls is only one issue amid other errand FDA priorities and perpetual personnel turnover, compounding and distracting from other critical strategic FDA reforms far more difficult — at precisely the moment they are most needed.
The FDA needs to restore its historical, robust, comprehensive, in-person inspection methodology. It must increase transparency to Americans regarding inspection failures and quality concerns. It must actively prioritize independent verification over manufacturer self-reporting and ensure that enforcement actions occur before dangerous products reach American patients — not just passively “tweet” about it on social media after the recall has taken place. Of note, that is currently what the FDA is doing, and has done over 7,400 times.
Drug safety and quality cannot operate on submissions and assumptions that are wrought with conflicts of interest relative to profit. Americans should not have to wonder whether medications from their pharmacies were manufactured under conditions the FDA has verified in a meaningful manner, nor should they be worried about device failure due to poor manufacturing practices. The FDA needs to eliminate its blind spots and stop relying on blind faith and trusting that overseas manufacturers are following the rules via remote inspections.
The FDA possesses the legal authority, institutional framework, and technical expertise necessary to solve this problem. What is missing is leadership’s willingness to demand the standards that once defined American drug regulation and legacy records of a low number of drug and device recalls.
FDA leadership must immediately return to a valid inspection process that made the FDA’s historical quality verification the global gold standard. FDA leadership and their inspectors need to get back to work and do so quickly.
AUTHOR DISCLAIMER: This article is not medical advice. Do NOT start or discontinue ANY drug without first discussing it with a pharmacist or physician you know and trust.
Author
